Ви дізнаєтеся, що таке хвороба Брутона (агаммаглобулинемия), її діагностика та лікування. Також розберемо генетичний профіль, основні ознаки і симптоми проблеми.
Що таке хвороба Брутона (агаммаглобулинемия)
Люди, у яких є цей розлад, мають низькі рівні захисних антитіл. Крім цього, вони уразливі до повторних і потенційно смертельним інфекцій.
Імунна система – це невід’ємний аспект здатності організму протистояти інфекціям мікроорганізмів (бактерій, вірусів, паразитів, грибів). Вона складається зі спеціалізованих клітин. Їх функція полягає в розпізнаванні чужих організмів і їх знищення.
Одними із спеціалізованих клітин, які використовуються для боротьби з інфекцією, є В-клітини . Вони циркулюють в кровотоці і продукують білки, що викликають боротьбу з організмом, звані антитілами.
Антитіла складаються з різних класів імуноглобулінів, які продукуються в В-клітці і потім вивільняються в кровотік. Там вони прикріплюються до вторгаються мікроорганізмів.
Є антитіла, спеціально призначені для об’єднання з кожним мікроорганізмом, дуже схожим на замок і ключ.
Як тільки антитіла приєднуються до мікроорганізму, він запускає інші спеціалізовані клітини імунної системи, щоб атакувати і знищувати загарбника. Тим самим йде боротьба з існуючою інфекцією.
Для того, щоб антитіла вироблялися організмом, В-клітини повинні розвиватися і дозрівати. Так вони можуть продукувати протиінфекційні антитіла.
Коли цей процес не відбувається нормально, імунна система не може добре працювати, щоб боротися з інфекцією. У підсумку, з’являється такий стан, відоме як імунодефіцит.
Аномалія в цьому розладі знаходиться в тирозинкіназ. Фермент, необхідному для дозрівання В-клітин. В результаті люди з цим станом мають низький рівень зрілих В-клітин і антитіл, які вони продукують, роблячи їх вразливими для частих і іноді небезпечних інфекцій.
Агаммаглобулінемія була першим захворюванням, викликаним імунодефіцитом, про який повідомив лікар Огден К. Брутон в 1952 році.
Пацієнт Брутона, чотирирічний хлопчик, вперше був госпіталізований у військовий госпіталь Уолтера Ріда через інфіковану коліна. Дитина одужала, коли Брутон дав йому антибіотики. Однак протягом наступних чотирьох років у нього було кілька інфекцій.
Генетичний профіль агаммаглобулінемії Брутона
Агаммаглобулінемія Брутона успадковується Х-зчепленим рецесивним чином. Якщо у жінки є один змінений ген BTK, вона буде носієм і піддаватися ризику передачі зміненого гена своїм дітям.
Оскільки батьки передають тільки Y-хромосому своїм синам і Х-хромосому своїм дочкам, жоден з синів порушеного чоловіки не розвиває розлад. Але все дочки будуть носіями.
За мутацію відповідають мутації в гені для BTK (розташовані в Xq21.3-22).
Було ідентифіковано більш 250 різних мутацій в BTK. Вони розподілені майже рівномірно по всьому гену BTK.
Хоча цей аномальний ген може передаватися від батьків до дитини, в половині випадків дитина буде показувати хвороба, не маючи батька з мутантним геном. Це пов’язано з тим, що можуть виникати нові зміни в гені BTK. Це нова зміна потім може бути передано дітям постраждалої людини.
Демографія
Агаммаглобулінемія зустрічається у всіх расових групах з частотою від одного до п’яти тисяч чоловік до одного на 100 000 чоловік.
Ознаки та симптоми хвороби Брутона
Агаммаглобулінемія є дефектом в В-клітинах. Це призводить до зниження антитіл в крові і підвищеної вразливості до інфекції деякими типами бактерій і вірусами.
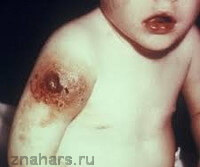
Діти з такою хворобою Брутона народжуються здоровими. Але вони починають проявляти ознаки інфекції в перші три-дев’ять місяців життя. Тобто в той час, коли зникають антитіла, які приходять від матері під час вагітності і раннього годування грудьми.
У 20-30% випадків пацієнти можуть мати більш високі рівні присутніх антитіл. Тоді симптоми можуть проявлятися пізніше.
Пацієнти можуть мати інфекції , пов’язані з:
- шкірою
- кістками
- мозком
- шлунково-кишковим трактом
- пазухами
- очима
- вухами
- носом
- дихальними шляхами в легені
- самим легким
Крім того, бактерії можуть мігрувати з початкового місця інфекції і потрапляти в кровотік. Це призводить до переважної зараження організму, що потенційно смертельно.
Крім ознак рецидивуючих інфекцій, у пацієнтів з агаммаглобулинемией можуть бути фізичні прояви :
- повільне зростання
- задишку
- невеликі мигдалини
- ненормальний рівень карієсу
У дітей можуть розвиватися такі незвичайні симптоми , як:
- захворювання суглобів
- руйнування еритроцитів
- ушкодження нирок
- запалення шкіри і м’язів
Збільшення захворюваності на рак, таким як лейкемія, лімфома і, можливо, рак товстої кишки, було пов’язано у невеликого відсотка людей.
Інфекції при агаммаглобулінемії Брутона
Інфекції при агаммаглобулінемії Брутона викликані бактеріями, які легко руйнуються від нормального функціонування імунної системи.
Поширені види бактерій :
- пневмокок
- стрептокок
- золотистий стафілокок
Хронічні захворювання шлунка і кишечника часто пов’язані з паразитом лямблії.
У хворих з хворобою Брутона організм може успішно захищати себе від вірусів і грибків, тому що інші аспекти імунної системи все ще функціонують.
Однак, є деякі помітні винятки!
Люди з цим розладом залишаються уразливими для вірусу гепатиту і вірусу поліомієліту. Віруси викликають особливу тривогу, так як це може призвести до прогресуючої та смертельної інфекції мозку, суглобів і шкіри.
- Діагностика рецидивуючих інфекцій або інфекцій, які не вдається повністю і швидко реагувати на антибіотики, повинні стати приводом для діагностичного пошуку для пацієнтів.
- Ще один ключ до діагностики агаммаглобулінемії є наявність незвично дрібних лімфатичних вузлів і мигдалин.
Крім того, багато пацієнтів з цим розладом мають історію тривалої хвороби. Тобто, вони не мають періоди благополуччя між нападами хвороби.
Коли пацієнт приходить з підозрою на хворобу Брутона, то діагноз встановлюється за допомогою декількох тестів.
Кількість імуноглобулінів вимірюється методом іммуноелектрофореза . При агаммаглобулінемії все імуноглобуліни будуть помітно знижені або відсутні.
Слід зазначити, що є деякі труднощі в діагностиці захворювання у немовляти або новонароджених. Адже імуноглобуліни від матері будуть з дитиною протягом перших кількох місяців життя.
Хворі також нездатні реагувати з утворенням антитіл після імунізації. Підтвердження діагнозу може бути зроблено шляхом демонстрації аномально низького кількість зрілих в-лімфоцитів в крові і генетичного дослідження, яке шукають мутації в гені БТК.
Коли діагноз даної хвороби підозрюється дитини, може бути запропоновано генетичне дослідження гена БТК. Це потрібно щоб визначити, чи є певні зміни генів, які можуть бути ідентифіковані. Якщо зміни виявлені, тестування може бути запропоновано для матері і родичок.
Пренатальна діагностика проводиться на клітинах, отриманих шляхом амніоцентезу (висновок рідини, що оточує плід в матці за допомогою голки) приблизно на 16 – 18 тижнях вагітності або з ворсин хоріона (частини плаценти).
У деяких сім’ях, зміни генів не можуть бути ідентифіковані.
Лікування хвороби Брутона
Поточні дослідження по лікуванню хвороби Брутона зосереджені на можливості трансплантації кісткового мозку або генної терапії для корекції аномального гена BTK. Однак в даний час лікування немає.

Тому є основні цілі:
- ефективно лікувати інфекцію
- запобігати повторні інфекції
- запобігати пошкодженню легких
Основний аномалією у пацієнтів з агаммаглобулинемией є недолік імуноглобулінів, які є будівельними блоками антитіл. Таким чином, лікування фокусується на заміну імуноглобуліну, тим самим забезпечуючи пацієнтів антитілами, необхідними для боротьби з інфекцією.
Імуноглобулін може бути отриманий з крові декількох донорів і переданий пацієнту з даною хворобою. Лікування імуноглобуліном проводиться кожні три-чотири тижні. Воно ефективно для профілактики інфекції різними мікроорганізмами.
Побічні ефекти або алергічні реакції на імуноглобулін нечасті. Але близько 3-12% людей відчувають задишку, підвищену пітливість, прискорене серцебиття, біль в шлунку, лихоманку, озноб, головний біль або нудоту.
Ці симптоми зазвичай слабшають, якщо імуноглобулін вводиться повільно, або реакції можуть зникнути після отримання імуноглобуліну кілька разів. Якщо реакції тривають, може знадобитися спеціальний процес фільтрації перед тим, як дати імуноглобулін пацієнтові.
Якщо інфекція відбувається у пацієнта з агаммаглобулинемией Брутона, антибіотики ( ліки, які вбивають бактерії ) можуть бути дані й, щоб допомогти боротися з інфекцією.
Періодичні або хронічні інфекції будуть розвиватися у деяких пацієнтів, незважаючи на використання імуноглобуліну. В цьому випадку антибіотики можна давати щодня, навіть якщо немає інфекції. Це необхідно щоб запобігти утворенню нової інфекції.
Якщо у пацієнта виникає хронічна діарея , необхідно провести тести, щоб знайти паразита Giardia lamblia. Також слід призначити належні антибіотики, щоб убити організм.
Профілактичні методи також дуже важливі!
Дітям з агаммаглобулинемией слід терміново лікувати навіть незначні порізи і подряпини. Також їм потрібно вчитися уникати натовпу і людей з інфекціями.
Людям з цим розладом і членам їх сімей не слід робити щеплення, що містять живі організми. Наприклад, поліомієліт або вакцину проти кору, епідемічного паротиту та краснухи. Інакше людина заразиться захворюванням, яке вакцинація покликана запобігти.
Напрямок для генетичного консультування підходить для родичів-жінок, які шукають інформацію про їх статус носія, а члени сім’ї приймають репродуктивні рішення.
прогноз
Без лікування імуноглобуліном 90% пацієнтів з агаммаглобулинемией Брутона помруть у віці восьми років.
У більшості пацієнтів, які регулярно отримують імуноглобулін, прогноз досить хороший. Вони повинні мати можливість вести щодо нормальне дитинство і не повинні бути ізольовані для запобігання небезпечних інфекцій.
Слід заохочувати повноцінний і активний спосіб життя!
Тепер ви знаєте, що таке хвороба Брутона і як її лікують на даний момент. Також ми розглянули основні ознаки і симптоми такої проблеми. Ще й генетичний профіль торкнулися. Загалом, будьте здорові!
Агаммаглобулінемія (хвороба Брутона)
Агаммаглобулінемія ( хвороба Брутона ) – це група спадкових імунодефіцитів, що характеризуються низькою концентрацією антитіл в крові через відсутність певних лімфоцитів в крові і лімфі.
Антитіла – це білки (імуноглобуліни, (IgM), (IgG) і ін.), Які є критичними і ключовими компонентами імунної системи.
Вони необхідні, якщо імунна система бореться з бактеріями, вірусами та іншими сторонніми речовинами, які загрожують організму.
Спеціалізовані клітини-попередники, які виробляють гамма-глобуліни, не можуть розвиватися або функціонувати належним чином, що призводить до дефіциту числа зрілих лімфоцитів, званих В-клітинами.
Типи агаммаглобулінемії : X-зчеплення агаммаглобулінемії (ХСА), набагато більш рідкісна X-зчеплення агаммаглобулінемії з дефіцитом гормону росту (зареєстровано близько 10 випадків) і аутосомно-рецесивна агаммаглобулинемия (ARAG). Всі ці розлади характеризуються ослабленою імунною системою, яку постійно потрібно посилювати введенням ін’єкцій гамма-глобуліну для боротьби з різними інфекціями.
симптоми агаммаглобулінемії
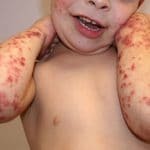
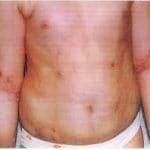
Рецидивуючі інфекції починають розвиватися в ранньому дитинстві і зберігаються протягом усього дорослого життя.
Найчастішим проявом хвороби Брутона або агаммаглобулінемії є збільшення сприйнятливості до інкапсульованим гнійним бактеріям , до таких як гемофільні інфекції, і деякі види Pseudomonas.
Шкірні інфекції у пацієнтів з хворобою в основному викликаються стрептококами групи А і стафілококами, вони можуть проявлятися у вигляді імпетиго , целюліту , абсцесів , або фурункулів .
Форма екземи, яка нагадує атопічний дерматит може бути очевидною, поряд зі збільшенням числа випадків гангренозний піодермії , вітіліго , алопеція і синдрому Стівенса-Джонсона (через більш широкого використання препаратів). Інші інфекції, які зазвичай присутні при цій хворобі, включають ентеровірусні інфекції, сепсис, менінгіт і бактеріальний пронос.
Пацієнти також можуть мати аутоімунні захворювання, тромбоцитопенії , нейтропенії , гемолітичні анемії і ревматоїдний артрит .
Постоянниеентеровірусние інфекції дуже рідко призводять до смертельного енцефаліту або синдрому дерматомиозит-менінгоенцефаліт.
На додаток до неврологічних змін, клінічні прояви цього синдрому включають набряки і еритематозну висип на шкірі над розгинальні суглобами.
Чоловіки, можуть розвивати незвично важкий і / або періодичний середній отит і пневмонії. Найбільш поширеним патогеном є S пневмонія, а потім вірус грипу B, стафілококи, менінгококи і моракселла катараліс.
У дітей у віці до 12 років, типові інфекції викликаються інкапсульованими бактеріями. Загальні інфекції в цій віковій групі включають рецидивні пневмонії, синусит, середній отит, які викликаються S пневмонія і вірусом грипу В, які важко піддаються лікуванню в такому віці.
У зрілому віці, шкірні прояви стають більш поширеними, як правило, через стафілокок і стрептокока групи А. Середній отит замінюється на хронічний синуситом, і хвороба легенів стає постійною проблемою, як в обмежувальної формі так і в обструктивної формі.
Як немовлята так і дорослі можуть мати аутоімунні захворювання. Як правило, ці розлади включають артрит, аутоімунні гемолітичні анемії, аутоімунних тромбоцитопенія, аутоімунні нейтропенії і запальні захворювання кишечника.
Запальні захворювання кишечника можуть бути дуже важко контрольованими і вони часто сприяють розвитку хронічної втрати ваги і недоїдання . Діарея є спільною і викликається лямбліями або кампілобактеріозом.
Пацієнти схильні до ентеровірусних інфекцій, в тому числі до поліовірусу.
причини
Агаммаглобулінемія – це рідкісне захворювання, яким в основному хворіють чоловіки. Провокується дана недуга генетичним дефектом, в результаті якого блокується зростання нормальних, сформованих імунних клітин – B-клітини (B-лімфоцити).
В результаті організм виробляє дуже маленька кількість (а іноді і зовсім не виробляє) імуноглобулінів в крові. Імуноглобуліни – відіграють важливу роль у захисті організму від різноманітних вірусів та інфекцій.
Люди, які страждають від агаммаглобулінемії, часто хворіють і особливо схильні до дії таких бактерій як: гемофільна паличка, пневмококи і стрептококи. Найчастіше супутні інфекції вражають:
- травний тракт;
- шкіру і суглоби;
- дихальні шляхи.
Хвороба Брутона – захворювання, що передається у спадок, а це означає, що аналогічна патологія може спостерігатися у інших родичів хворого.
Ускладнення при агаммаглобулінемії
Ускладнення включають хронічні інфекції, ентеровірусні інфекції центральної нервової системи, збільшення частоти розвитку аутоімунних захворювань, а також інфекції шкіри. Пацієнти мають підвищений ризик розвитку лімфоми.
діагностика
Раннє виявлення та діагностика має важливе значення для запобігання ранньої захворюваності і смерті від системних і легеневих інфекцій.
Діагноз підтверджується аномально низькими рівнями або взагалі відсутніми зрілими В-лімфоцитами, а також низькою або відсутньою експресією важкого ланцюга м на поверхні лімфоцитів.
З іншого боку, рівень Т-лімфоцитів буде підвищеним. Остаточний визначник хвороби – молекулярний аналіз.
Молекулярний аналіз також використовується для пренатальної діагностики, яка може бути виконана за допомогою відбору проб ворсинок хоріона або амніоцентезу, коли мати, як відомо, є носієм дефектного гена. Рівні IgG менше 100 мг / дл підтверджують діагноз.
Рідко, але діагноз може бути поставлений у дорослих в їх другому десятилітті життя. Це, як вважають, відбувається через мутації в білку, а не через його повної відсутності.
лабораторні аналізи
На першому етапі необхідно провести кількісний вимір IgG, IgM, імуноглобуліну Е (IgE) і імуноглобулін А (IgA). Рівні IgG слід вимірювати по-перше, бажано після віку 6 місяців, коли рівні материнських IgG почнуть знижуватися. По-друге, рівні IgG нижче 100 мг / дл, як правило, свідчить про хворобу Брутона. Як правило, IgM і IgA не виявляються.
Після того, як рівень антитіл буде визначено як аномально низький, підтвердження діагнозу буде досягнуто за допомогою аналізу B-лімфоцітний і Т-лімфоцітний маркерів. Рівні CD19 + В-клітин нижче 100 мг / дл. Значення аналізу Т-клітин (CD4 + і CD8 +), як правило, збільшуються.
Подальший аналіз може бути проведений шляхом виявлення відповідей IgG до Т-залежним і Т-незалежним антигенів, шляхом проведення імунізації, наприклад після введення некон’югованій 23-валентної пневмококової вакцини або вакцин від дифтерії, правця і H грипу типу B.
Молекулярно-генетичним дослідженням можна встановити раннє підтвердження діагнозу вродженої агаммаглобулінемії.
інші обстеження
Дослідження функцій легких займають центральне місце в моніторингу захворювань легенів. Вони повинні проводитися щорічно у дітей, які можуть виконувати тест (зазвичай від 5 років).
процедури
Ендоскопія і колоноскопія можуть бути використані для оцінки масштабів і прогресування запального захворювання кишечника. Бронхоскопія може бути корисною в діагностиці та відстеження хронічного захворювання легенів і інфекцій.
лікування агаммаглобулінемії

Замісна терапія гамма-глобуліном є стандартним методом лікування агаммаглобулінемії. Внутрішньовенне введення гамма-глобулін використовується для лікування агаммаглобулінемії і загального вариабельного імунодефіциту.
Антибіотики призначаються людям з агаммаглобулинемией при бактеріальних інфекціях. Деяких пацієнтів лікують антибіотиками в якості профілактичного заходу (профілактично).
Всі люди з імунодефіцитом повинні бути максимально захищені від впливу інфекційних захворювань.
Слід по можливості уникати кортикостероїдів або будь-яких ліків , які пригнічують імунну систему (імунодепресанти), а також таких фізичних навантажень, як грубі контактні види спорту, які можуть пошкодити селезінку.
Як вже зазначалося вище, у людей з імунодефіцитом з підвищеним рівнем IgM спостерігається схильність до кровотеч, надмірно пов’язаних з аномально низьким рівнем циркулюючих тромбоцитів в крові (тромбоцитопенія). Це може ускладнити будь-яку хірургічну процедуру.
прогноз
Агаммаглобулінемія має сприятливий прогноз за умови постійного введення гамма-глобуліном і здійснення адекватної терапії антибіотиками.
Несвоєчасне застосування антибактеріальної терапії при загостренні інфекційних хвороб може призвести до стрімкого прогресування патологічного процесу і летального результату хворого.
агаммаглобулінемія

Агаммаглобулінемія – це спадково обумовлене захворювання, при якому розвивається важкий первинний імунодефіцит (дефект імунного захисту організму) з вираженим зниженням рівня гамма-глобулінів в крові. Недуга проявляється зазвичай у перші місяці і роки життя дитини, коли починають розвиватися повторні бактеріальні інфекції: отит, синусит, пневмонії, піодермії, менінгіт, сепсис.
При обстеженні в периферичної крові та кістковому мозку практично відсутні сироваткові імуноглобуліни і B-клітини. Лікування агаммаглобулінемії полягає в довічної замісної терапії.
Агаммаглобулінемія (спадкова гіпогамаглобулінемія, хвороба Брутона) – вроджений дефект гуморального імунітету, обумовлений мутаціями в геномі клітин, що призводить до недостатності синтезу B-лімфоцитів.
В результаті порушується утворення імуноглобулінів всіх класів, і їх вміст у крові різко знижується аж до повної відсутності, розвивається первинний імунодефіцит. Низька реактивність імунної системи призводить до розвитку важких повторних гнійно-запальних захворювань ЛОР-органів, бронхів і легенів, шлунково-кишкового тракту і мозкових оболонок.
Хвороба Брутона зустрічається виключно у хлопчиків і спостерігається приблизно у 1-5 чоловік з мільйона новонароджених, незалежно від раси та етнічної групи.
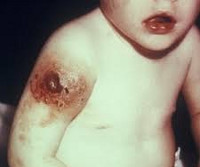
X-зчеплена форма спадкової агаммаглобулінемії виникає внаслідок пошкодження одного з генів X-хромосоми (розташований на Xq21.3-22.2). Цей ген відповідальний за синтез ферменту тирозинкінази, який бере участь в процесі освіти і диференціювання B-клітин. В результаті мутацій цього гена і блокування синтезу брутоновской тирозинкінази порушується формування гуморального імунітету.
При агаммаглобулінемії молоді форми (пре-B-клітини) присутні в кістковому мозку, а їх подальша диференціювання і надходження в кровоносне русло порушена. Відповідно, вироблення всіх класів імуноглобулінів практично не проводиться, і організм дитини стає беззахисним при проникненні хвороботворних бактерій (найчастіше це стрептококи, стафілококи і синьогнійна паличка).
Подібний механізм порушень відзначається і у випадку з іншою формою спадкової агаммоглобулінеміі – зчепленої з X-хромосомою і недостатністю гормону росту. Аутосомно-рецесивна форма розвивається в результаті мутації декількох генів (μ-важких ланцюгів, гена λ5 / 14.1, гена Адапторная білка і гена сигнальної молекули IgА).
Виділяють три форми спадкової агаммаглобулінемії:
- сцепленная з X-хромосомою (85% всіх випадків вроджених гіпогаммаглобулінемій, хворіють тільки хлопчики)
- аутосомно-рецесивна спорадична швейцарського типу (зустрічається у хлопчиків і дівчаток)
- сцепленная з X-хромосомою і недостатністю гормону росту агаммаглобулинемия (зустрічається вкрай рідко і тільки у хлопчиків)
Знижена реактивність гуморального імунітету при агаммаглобулінемія призводить до розвитку повторних гнійно-запальних захворювань вже на першому році життя дитини (як правило, після припинення грудного вигодовування – в 6-8 місяців).
При цьому захисні антитіла від матері в організм дитини вже не надходять, а свої імуноглобуліни – які не виробляються. До 3-4 річного віку запальні процеси переходять у хронічну форму зі схильністю до генералізації.
Гнійна інфекція при агаммаглобулінемії може вражати різні органи і системи.
З боку ЛОР-органів нерідкі гнійні гайморити, етмоїдити, отити, причому гнійний отит частіше розвивається на першому році життя дитини, а синусити – в 3-5 років. Із захворювань бронхолегеневої системи спостерігаються повторні бронхіти, пневмонії, абсцеси легкого.
Часто зустрічається ураження шлунково-кишкового тракту з наполегливої діареєю (проносами), викликаної хронічним інфекційним ентероколіт (основні збудники – кампілобактерії, лямблії, ротавірус). На шкірних покривах виявляється імпетиго, мікробна екзема, рецидивуючий фурункульоз, абсцеси і флегмони.
Нерідко буває ураження очей (гнійні кон’юнктивіти), порожнини рота (виразкові стоматити, гінгівіти), кістково-м’язової системи (остеомієліти, гнійні артрити).
В цілому клінічна картина агаммаглобулінемії характеризується поєднанням загальних симптомів, які спостерігаються при гнійної інфекції (висока температура тіла, озноб, болі в м’язах і головний біль, загальна слабкість, порушення сну і апетиту і т. Д.) І ознак ураження конкретного органу (кашель, задишка , утруднення носового дихання, гнійні виділення, діарея і т. п.).
Будь-яке інфекційне і соматичне захворювання у хворого імунодефіцитом протікає важко, довго і супроводжується ускладненнями.
Важкий перебіг агаммоглобулінеміі може ускладнюватися розвитком менінгіту, вірусного енцефаломієліту, поствакцинального паралітичного поліомієліту, сепсису.
На тлі захворювання підвищена ймовірність розвитку аутоімунних і онкологічних захворювань. Загибель пацієнтів часто настає від інфекційно-токсичного шоку.
При клінічному огляді пацієнта лікарем алергологом-імунологом виявляються ознаки гнійно-запального ураження того чи іншого органу (тканини) і симптоми, що підтверджують знижену реактивність імунної системи: гіпоплазія мигдалин, зменшення периферичних лімфатичних вузлів. Виражені і ознаки відставання у фізичному розвитку дитини.
Лабораторне дослідження крові виявляє виражене зниження рівня імуноглобулінів в иммунограмме (IgA і IgM <20 мг / дл – зниження в сто раз, IgG <200 мг / дл – у десять разів) або їх повна відсутність.
У периферичної крові виявляється глибокий дефіцит B-клітин (менше 1%), в кістковому мозку практично відсутні плазмоцити. Що стосується клітинного імунітету, то зміст T-лімфоцитів може бути в нормі.
Під час гістологічного дослідження лімфоїдної тканини гермінативні (зародкові) центри і плазматичні клітини відсутні.
Диференціальна діагностика спадкової агаммаглобулінемії проводиться з іншими первинними і вторинними імунодефіцитними станами (генетичними порушеннями, ВІЛ і цитомегаловірусом, вродженою краснухою і токсоплазмозом, злоякісними новоутвореннями і системними порушеннями, імунодефіцитом внаслідок інтоксикації лікарськими препаратами та ін.).
Необхідна довічна замісна терапія антітелосодержащімі препаратами. Зазвичай використовується введення внутрішньовенного імуноглобуліну, а при його відсутності – нативної плазми від здорових постійних донорів.
При вперше встановленого діагнозу агаммаглобулінемії заместительное лікування проводиться в режимі насичення до досягнення рівня імуноглобуліну IgG вище 400 мг / дл, після чого при відсутності активного гнійно-запального процесу в органах і тканинах можна переходити до підтримуючої терапії з введенням профілактичних доз препаратів, що містять імуноглобуліни.
Будь-епізод бактеріальної гнійної інфекції, незалежно від локалізації запального процесу, вимагає проведення адекватної антибактеріальної терапії, яка виконується одночасно з замінних лікуванням.
Найчастіше при агаммаглобулінемії використовуються антибактеріальні засоби з групи цефалоспоринів, аміноглікозидів, макролідів, а також антибіотики пеніцилінового ряду.
Тривалість лікування при цьому в кілька разів перевищує стандартну при даному захворюванні.
Симптоматичне лікування проводиться з урахуванням конкретного ураження того чи іншого органу (промивання навколоносових пазух носа антисептиками, виконання вібраційного масажу грудної клітки і постурального дренажу при бронхітах і пневмоніях і т. Д.).
Якщо агаммаглобулинемия виявлена в ранньому віці до настання важких ускладнень, і адекватна стану пацієнта замісна терапія почата вчасно, можливо збереження нормального способу життя протягом багатьох років.
Однак в більшості випадків діагностика спадкових порушень гуморального імунітету здійснюється занадто пізно, коли вже розвинулися незворотні хронічні гнійно-запальні захворювання органів і систем організму.
У цьому випадку прогноз при агаммаглобулінемії несприятливий.
Агаммаглобулінемія у дітей
Агаммаглобулінемія – спадкове захворювання, що характеризується відсутністю або різким зниженням вмісту в крові γ-глобулінів.
Незважаючи на те, що агаммаглобулинемия – рідкісне захворювання (частота становить 1: 1 000 000), вона буває найбільш поширеною формою первинного імунодефіциту .
Тип успадкування – рецесивний, зчеплений з X-хромосомою. Зустрічають тільки у хлопчиків .
Встановлено, що ген, відповідальний за розвиток Х-зчепленої агаммаглобулінемії, кодує білок
тирозинкіназ. При цій формі імунодефіциту відсутні В-клітини і плазмоцити, в лімфоїдної тканини немає центрів розмноження, в сироватці не вдається виявити IgM,
IgD, IgA або IgE (Ig – імуноглобулін).
IgG вкрай низька: складає всього 10% норми. У той же час хворі мають нормально розвинену вилочкової залози. Т-клітини в кількісному і функціональному відношенні не відрізняються від таких у здорових дітей.
Починається захворювання зазвичай з 2-го року життя, але перші його ознаки
можуть з’явитися вже в кінці першого року.
До 9-12-місячного віку хвороба не проявляється, тому що новонароджені
отримують захисні антитіла з молоком матері.
Однак після цього періоду дитина піддається постійному інфікування такими збудниками, як Haemophilus influenzae, Streptococcus pneumoniae, Staphylococcus aureus.
Клінічно агаммаглобулинемия характеризується рецидивуючими бактеріальними інфекціями, в той час як вірусні зазвичай хворі переносять задовільно.
Інфекційні хвороби при агаммаглобулінемії протікають тривало, з частими загостреннями і важкими ускладненнями. Виникають повторні пневмонії, отити, піодермії, сепсис.
діагностика агаммаглобулінемії
Критерієм діагнозу є зниження концентрація сироваткового IgG менше 2 г / л при відсутності IgA, IgM та циркулюючих B-лімфоцитів. У деяких хворих з XLA відзначається «аттенуіровані» фенотип, при якому можуть виявлятися слідові кількості IgG і IgM, і число В лімфоцитів в периферичної крові становить до 0,5% всіх лімфоцитів.
Зазвичай такі хворі мають більш пізній дебют захворювання.
Низький відсоток (майже відсутність) В-клітин в крові є найхарактернішим і надійним лабораторним ознакою Х-СА.
Якщо брат новонародженого хлопчика, або двоюрідний або рідний брат матері страждає агаммаглобулинемией, цей новонароджений має ризик агаммаглобулінемії і його сім’я і лікарі повинні негайно визначити відсоток В-клітин в крові, щоб почати лікування до того, як дитина захворіє.
Діагноз агаммаглобулинемия може бути підтверджений відсутністю білка BTK в моноцитах або тромбоцитах, або виявленням мутації BTK в ДНК. Майже кожна сім’я має свою особливу мутацію BTK, проте члени однієї і тієї ж сім’ї зазвичай мають одну і ту ж мутацію.
Спадкування агаммаглобулінемії
Х-зчеплення агаммаглобулінемії (Х-СА) є генетичним захворюванням, яке може успадковуватися і бути сімейним. Воно успадковується як зчеплена з Х-хромосомою рецесивний ознака.
Важливо знати тип спадкування, щоб члени сім’ї краще розуміли, чому захворіла дитина, який ризик захворювання у наступних дітей, і яке це має значення для інших членів сім’ї.
Після ідентифікації гена, що викликає Х-СА, стало можливим досліджувати сестер пацієнта з Х-СА та інших членів сім’ї жіночої статі, наприклад, рідних сестер матері дитини, щоб визначити, чи не є вони носіями цього захворювання.
У носіїв Х-СА захворювання не проявляється, проте вони можуть передати його своїм синам з імовірністю 50%. У деяких випадках також можливо виявити Х-СА у плода до народження. Зараз ці генетичні дослідження проводяться лише в декількох лабораторіях.
лікування агаммаглобулінемії
Основою лікування згаммаглобулінеміі є замісна терапія внутрішньовенним імуноглобуліном. В даний час не існує методів лікування пацієнтів з Х-зчепленої агаммагло-булінеміей (Х-СА).
Дефектний ген не можна виправити або замінити, а дозрівання попередників В-лімфоцитів в В-лімфоцити і плазматичні клітини неможливо стимулювати.
Однак хворим з Х-зчепленої агаммаглобулинемией можна вводити деякі відсутні у них антитіла. Ці антитіла є у вигляді імуноглобулінів (або гамма-глобулінів) і можуть бути введені безпосередньо в кров (внутрішньовенно) або під шкіру.
Препарати імуноглобулінів містять антитіла, які замінять антитіла, які організм хворого Х-СА не може виробляти самостійно. Вони містять антитіла до широкого спектру мікроорганізмів.
Імуноглобуліни особливо ефективні в профілактиці поширення інфекції в кров і глибокі внутрішні органи або тканини. Деяким пацієнтам допомагає щоденний прийом антибіотиків всередину для захисту від інфекції, або для лікування хронічного синуситу або бронхіту.
Пацієнти з Х-зчепленої агаммагло-булінеміей не повинні піддаватися вакцинації на основі живих вірусів, наприклад, живий противопол-міелітной вакциною, а також одержала вакциною від кору, свинки і краснухи (КСК).
Існує невелика ймовірність того, що живі вакцини (особливо пероральна протівополіоміелітная вакцина) можуть у хворих агаммагобулінеміей стати джерелом тих захворювань, для профілактики яких вони розроблені.
При наявності хронічних вогнищ інфекції (як правило, в легких) хворим показано призначення постійної профілактичної антибактеріальної терапії. У разі наполегливої нейтропенії, яка супроводжується клінічними проявами застосовують ростові фактори. Трансплантація стовбурових клітин при агаммаглобулінемії не відображено.
Прогноз при агаммаглобулінемії
Застосування комбінованої терапії агаммаглобулінемії з використанням регулярних введень імуноглобуліну і антибіотиків значно поліпшило прогноз захворювання.
Рано розпочата адекватна замісна терапія внутрішньовенним імуноглобуліном дозволяє уникнути формування хронічних інфекцій і значно зменшує кількість епізодів гострих інфекцій і частоту розвитку інших ускладнень. Останнім часом, більшість пацієнтів, які отримують адекватне лікування, досягають дорослого віку.
Більшість пацієнтів з Х-зчепленої агаммаглобулинемией (Х-СА), регулярно одержують імуноглобуліни, здатні вести відносно нормальне життя. Вони не потребують ізоляції або обмеження діяльності. Слід заохочувати активне заняття командними видами спорту.
Час від часу інфекції можуть вимагати особливої уваги, проте діти з Х-зчепленої агаммагло-булінеміей можуть брати участь у всіх шкільних і позашкільних заходах, а при досягненні дорослого віку вони можуть продуктивно працювати і мати сім’ю. Слід налаштовувати дитину на повністю активний стиль життя і заохочувати його!
Первинний імунодефіцит. Х-зчеплення агаммаглобулінемії
Х-зчеплення агаммаглобулінемії (Х-СА) була вперше описана в 1952 році д-ром Огден Брутона. Це захворювання, яке іноді називають агаммаглобулинемией Брутона або врожденнойагаммаглобулінеміей, було одним з перших виявлених імунодефіцитних захворювань.
Х-СА є спадковим захворюванням, при якому організм пацієнта не способенвирабативать антитіла, білки, з яких складається гамма-глобулінової або иммуноглобулинового фракція плазми крові. Антитіла є невід’ємною частиною механізму захисту організму проти деяких мікроорганізмів (наприклад, бактерій і вірусів). Антитіла необхідні для одужання від інфекцій.
Вони також захищають від повторного захворювання некоториміінфекціямі. Існують антитіла, спеціально призначені для з’єднання з конкретним мікроорганізмом – це схоже на відповідність ключа замку. Коли такі мікроорганізми, як бактерії, потрапляють на слизові оболонки або всередину організму, молекули антитіл, спеціфіч- них для даного мікроорганізму, прилипають до його поверхні.
Прикріплення антитіла до поверхні мікроорганізму може мати один або кілька ефектів, корисних для людини. Наприклад, деякі мікроорганізми повинні прикріпитися до клітин організму для того, щоб викликати інфекцію, а антитіла не дають мікроорганізмам “приклеюватися” до клітин.
Антитіла, прикріплені до поверхні деяких мікроорганізмів, також активують інші захисні сили організму (наприклад, групу білків крові, яка називається сироватковим комплек-ментом), які можуть безпосередньо знищувати бактерії або віруси.
Нарешті, бактерії, покриті антитілами, набагато легше проковтують і знищуються білими клітинами крові (фагоцитами), ніж бактерії, не покриті антитілами. Всі ці механізми не дають мікроор-організми проникати в тканини організму, де вони можуть викликати важкі інфекціі.Основним дефектом при Х-СА є нездатність пацієнта виробляти антитіла.
Антитіла – це білки, що виробляються спеціальними клітинами, які називаються плазматичними клітинами. Розвиток плазматичних клітин проходить в определенномпорядке, починаючи зі стовбурових клітин, розташованих в кістковому мозку. З стовбурових клітин утворюються незрілі лімфоцити, звані про-В-лімфоцитами.
Про-В-лімфоцити являютсяпредшественнікамі пре-В-лімфоцитів, з яких в свою чергу утворюються В-лімфоцити. Кожен По-лімфоцит несе на своїй поверхні зразок імуноглобуліну, який ця клітина може виробляти. Цей поверхневий клітинний імуноглобулін може зв’язуватися з чужорідними речовинами, званими антигенами.
Коли В-лімфоцит контактує зі своїм специфічним антигеном, наприклад, пневмококком або правцевим анатоксином, він дозріває в плазматичну клітину, що виділяє антитіла. Кожна В-клітина виробляє антитіло (імуноглобулін), злегка відрізняється від продуктів інших клітин, що дозволяє організму відповідати на мільйони різних сторонніх речовин.
У більшості хворих Х-СА є попередники В-лімфоцитів, але далеко не всі з них потім стають В-лімфоці- тами. В результаті цього при Х-СА основним дефектом є нездатність предшествен- ників В-лімфоцитів дозрівати до стану В-лімфоцитів.
Хворі з Х-СА мають мутації в гені, необхідному для нормального розвитку В-лімфоцитів. Цей ген, відкритий в 1993 році, називається BTK (ген тирозинкінази Брутона) в честь першовідкривача цього порушення – полковника, д-ра медицини Огдена Брутона. Як показує назва цього порушення, ген BTK розташований в Х-хромосомі.
Клінічні прояви
Хворі Х-зчепленої агаммаглобулинемией (Х-СА) схильні до інфекцій у зв’язку з недос-татка антитіл.
Ці інфекції часто розвиваються на поверхні або поблизу поверхні слизових оболонок, наприклад, в середньому вусі, придаткових пазухах і легких, проте в деяких випадках інфекція може вражати циркулює кров або внутрішні органи.
В результаті цього у хворих з Х-СА розвиваються інфекції придаткових пазух носа (синусит), очей (кон’юнктивіт), вух (отит), носа (риніт), внутрілегочних дихальних шляхів (бронхіт) або самих легких (пневмонія). Можлива також інфекція шлунково-кишкового тракту, в част- ності, викликана кишковою лямблій Giardia.
Кишкова лямблії може викликати біль в животі, пронос, уповільнення зростання або втрату білків крові, наприклад, гамма-глобуліну. Некоториепаціенти з Х-СА також схильні до шкірних інфекцій.
При відсутності антитіл будь-яка з цих інфекцій може потрапити в кровотік і поширитися на інші органи вглиб організму, наприклад, на кістки, суглоби або головний мозок. У пацієнтів з Х-СА інфекції зазвичай викликаються мікроорганізмами, які дуже швидко знищуються або інактивуються антитілами у здорових людей.
Збудниками таких інфекцій найчастіше є пневмокок, стрептокок, стафілокок або Haemophilus influenzae. Деякі особливі види вірусів також можуть стати причиною важкої інфекції у цих пацієнтів.
При фізикальному обстеженні у більшості хворих з Х-СА виявляють дуже маленькі мигдалини і лімфатичні вузли (шийні залози). Це пов’язано з тим, що мигдалини і лімфатичні вузли в основному складаються з В-лімфоцитів. При відсутності В-лімфоцитів ці тканини зменшуються.
діагноз
Діагноз Х-СА слід мати на увазі при обстеженні будь-якого хлопчика з рецидивуючими або важкими бактеріальними інфекціями, особливо при невеликому розмірі або отсутствііміндалін і лімфатичних вузлів. Першим дослідженням з метою скринінгу повинно бути визначення імуноглобулінів сироватки крові.
У більшості випадків при Х-СА виявляється значне зниження або відсутність імуноглобулінів (IgG, IgM і IgA). Однак бувають і винятки: у деяких пацієнтів зберігаються деякі кількості IgM або IgG.
Крім того, новонароджені протягом перших місяців життя в нормі вирабати ють лише невеликі кількості імуноглобулінів, в зв’язку з чим іноді важко відрізнити новонародженого з нормальною затримкою вироблення імуноглобулінів від новонародженого з істинною імунною недостатністю.
Якщо рівень сироваткових імуноглобулінів низький або у лікаря є серйозна підозра, що пацієнт страждає Х-СА, слід визначити число В-клітин в периферійній крові. Низький відсоток (майже відсутність) В-клітин в крові є найхарактернішим і надійним лабораторним ознакою Х-СА.
Якщо брат новонародженого хлопчика, або двоюрідний або рідний брат матері страждає Х-СА, цей новонароджений має ризик Х-СА, і його сім’я і лікарі повинні негайно визначити відсоток В-клітин в крові, щоб почати лікування до того, як дитина захворіє.
Діагноз Х-СА може бути підтверджений відсутністю білка BTK в моноцитах або тромбоцитах, або виявленням мутації BTK в ДНК. Майже кожна сім’я має свою особливу мутацію BTK, проте члени однієї і тієї ж сім’ї зазвичай мають одну і ту ж мутацію.
спадкування
Х-зчеплення агаммаглобулінемії (Х-СА) є генетичним захворюванням, яке може успадковуватися і бути сімейним. Воно успадковується як зчеплена з Х-хромосомою рецесивний ознака.
Важливо знати тип спадкування, щоб члени сім’ї краще розуміли, чому захворіла дитина, який ризик захворювання у наступних дітей, і яке це має значення для інших членів сім’ї.
Після ідентифікації гена, що викликає Х-СА, стало можливим досліджувати сестер пацієнта з Х-СА та інших членів сім’ї жіночої статі, наприклад, рідних сестер матері дитини, щоб визначити, чи не є вони носіями цього захворювання.
У носіїв Х-СА захворювання не проявляється, проте вони можуть передати його своїм синам з імовірністю 50%. У деяких випадках також можливо виявити Х-СА у плода до народження. Зараз ці генетичні дослідження проводяться лише в декількох лабораторіях.
лікування
В даний час не існує методів лікування пацієнтів з Х-зчепленої агаммагло-булінеміей (Х-СА). Дефектний ген не можна виправити або замінити, а дозрівання предшест-венников В-лімфоцитів в В-лімфоцити і плазматичні клітини неможливо стимулювати.
Однак хворим з Х-СА можна вводити деякі відсутні у них антитіла. Ці антитіла є у вигляді імуноглобулінів (або гамма-глобулінів) і можуть бути введені безпосередній але в кров (внутрішньовенно) або під шкіру.
Препарати імуноглобулінів містять антитіла, які замінять антитіла, які організм хворого Х-СА не може виробляти самостійно. Вони містять антитіла до широкого спектру мікроорганізмів. Імуноглобуліни особенноеффектівни в профілактиці поширення інфекції в кров і глибокі внутрішні органи або тканини.
Деяким пацієнтам допомагає щоденний прийом антибіотиків всередину для захисту від інфекції, або для лікування хронічного синуситу або бронхіту. Пацієнти з Х-СА не повинні піддаватися вакцинації на основі живих вірусів, наприклад, живий противопол-міелітной вакциною, а також одержала вакциною від кору, свинки і краснухи (КСК).
Существуетнебольшая ймовірність того, що живі вакцини (особливо пероральна протівополіоміеліт- ная вакцина) можуть у хворих агаммагобулінеміей стати джерелом тих захворювань, для профілактики яких вони розроблені.
прогноз
Більшість пацієнтів з Х-зчепленої агаммаглобулинемией (Х-СА), регулярно одержують імуноглобуліни, здатні вести відносно нормальне життя. Вони не потребують ізоляції або обмеження діяльності. Слід заохочувати активне заняття командними видами спорту.
Час від часу інфекції можуть вимагати особливої уваги, проте діти з Х-СА можуть брати участь у всіх шкільних і позашкільних заходах, а при досягненні дорослого віку вони можуть продуктивно працювати і мати сім’ю. Слід налаштовувати дитину на повністю активний стиль життя і заохочувати його!
Стаття люб’язно надана всесвітньою організацією IPOPI – працює на поліпшення життя людей з первинним імунодефіцитом. Авторські права 2007 належать фонду Immune Deficiency Foundation, США.
«Керівництво по первинним імунодефіцитних захворювань для хворих і членів їх сімей», з якого цей матеріал взято за ліцензією, було розроблено Immune Deficiency Foundation за підтримки компанії Baxter Healthcare Corporation.