Зараз діагностується величезна кількість спадкових захворювань, які дитина отримує від батька або від матері.
Екологічна обстановка, неправильне харчування, нездоровий спосіб життя – все це призводить до того, що клітини мутують, генетична інформація зазнає суттєвих змін.
Ось від цього і виникає величезна кількість спадкових захворювань. Одним з них є фенілкетонурія. Що це за хвороба, знають не багато, тому постараємося в усьому розібратися.
сутність поняття
Фенілкетонурія є спадковим захворюванням, пов’язане воно з серйозними порушеннями в білковому обміні. Це, в свою чергу, призводить до ураження нервової системи.
Непрацездатність за все одного ферменту, фенілаланіну, а в результаті – такі серйозні проблеми зі здоров’ям, як фенілкетонурія. Що це за стан, коли в організмі відбувається накопичення великої кількості токсичних речовин? Всі отруйні сполуки запасаються в біологічних рідинах, тому для медиків діагностувати хворобу зазвичай не складає особливих труднощів.
- Якщо вчасно не вжити заходів, то можна спостерігати серйозні ураження нервової системи, а це вже призводить до порушень у функціонуванні всього організму.
- Таким чином, без відповідного лікування про нормальне життя пацієнта не може бути й мови.
причини захворювання
Всі білки складаються з амінокислот, яких всього 20, але серед них є такі, які синтезуються в організмі людини. Деякі ж повинні надходити тільки ззовні. До незамінних амінокислот відноситься і фенілаланін.
У здорової людини він, потрапляючи всередину, перетворюється в тирозин. Це абсолютно інша амінокислота, і тільки кілька відсотків речовини направляється в нирки і там перетворюється в фенілкетон, досить токсична речовина.
Якщо у людини відсутній фермент фенілаланін-4-гідроксилази або неправильно працює той, який перетворює фенілаланін в іншу речовину, то ось в цьому випадку і розвивається фенілкетонурія. Що це досить серйозний симптом, скаже вам кожен лікар, тому необхідно вживати термінових заходів.
А привести до відсутності потрібного ферменту може мутація гена в хромосомі 12.
різновиди фенілкетонурії
Якщо розглядати форми захворювання, то вони можуть бути такими:
- Класична. В цьому випадку ми спостерігаємо, що фенілкетонурія – рецесивний ознака. Зустрічається така форма у одну дитину на десять тисяч здорових дітей. Якщо не вжити заходів, то навряд чи хвора людина доживе до тридцяти років.
- Врятовано форма. Чи не передається у спадок, а викликає її мутація в генах. Перебіг її тяжче, а рання смертність прогнозується з ймовірністю практично 100%.

Крім форм, лікарі розрізняють ще й типи фенілкетонурії:
- Перший тип характеризується тим, що спостерігається нестача ферменту фенілаланін-4-гідроксилази, який і відповідає за перетворення фенілаланіну. У 98% випадків діагностується саме він.
- Другий. Його відрізняє малий вміст ферменту дігідроптерідінредуктази. У таких хворих спостерігаються судоми, а також розумова відсталість. Незважаючи на свою рідкісну зустрічальність, смертність від цього типу може наступати в 2-3-річному віці.
- Третій тип характеризується тим, що виникає дефіцит тетрагідробіоптеріна. В результаті відбувається зменшення обсягів мозку, що призводить до розумової відсталості.
симптоматика хвороби
Відразу після народження дитини за її зовнішнім виглядом або поведінкою важко діагностувати захворювання. Основні ознаки почнуть проявлятися трохи згодом. Однак ще в пологовому будинку медики цілком здатні поставити діагноз «фенілкетонурія». Симптоми у цього захворювання наступні:
- часта блювота без видимих причин;
- плаксивість;
- млявість;
- можуть з’являтися висипання по всьому тілу;
- сеча має «мишачий» запах;
- дитина відстає у фізичному та розумовому розвитку від своїх однолітків.
Досить взяти аналіз крові і сечі, щоб поставити правильний діагноз.
ознаки фенілкетонурії
Поступово при відсутності належного лікування у хворого можна буде спостерігати такі ознаки:
- Судомний синдром. Він починає проявлятися в ранньому дитинстві і зберігається у дорослих.
- Недолік пігменту в шкірі та волоссі. Тому такі пацієнти, як правило, світловолосі і мають білу шкіру.
- Запальні процеси, які через незнання можна прийняти за алергічну реакцію.

Перші ознаки розумового відставання можна помітити у дитини вже у віці півроку. Він перестає запам’ятовувати нову інформацію, і здається, що він абсолютно не здатний до навчання.
Бути пильними батьки повинні і тоді, коли малюк забуває те, чого вже давно навчився, наприклад, як тримати ложку, сидіти, грати з брязкальцем.
Тривогу потрібно бити і в тому випадку, якщо дитина перестає впізнавати батьків і близьких людей, а також надмірна плаксивість з віком не проходить.
Ось ознаки, які має фенілкетонурія, симптоми хвороби необхідно розглядати тільки в комплексі, тому що окремо вони цілком можуть зустрічатися і у здорових дітей.
виявлення захворювання
Поставити правильний діагноз можна двома шляхами:
- Провести аналіз крові і сечі у новонародженого ще в пологовому будинку. це, як правило, роблять у всіх випадках.
- Визначити наявність фенілкетонов в біологічних рідинах дорослої людини при наявності відповідних ознак.
У дітей в пологовому будинку кров беруть на 4-5-й день і визначають зміст фенілаланіну. Якщо такий виявляється, то дитину з мамою направляють на консультацію до генетика.
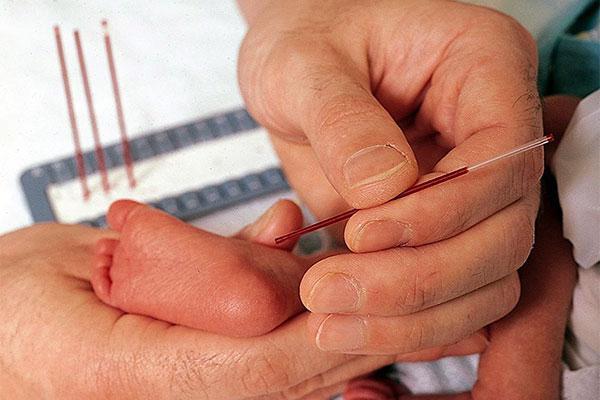
Перед випискою обов’язково поцікавтеся, чи робили вашій дитині аналіз на фенілкетонурію. Незважаючи на невелику зустрічальність цього захворювання, найкращим рішенням буде все одно підстрахуватися.
спадкування
Так як фенілкетонурія успадковується як рецесивна ознака, то для прояву його у дитини необхідно, щоб у обох батьків був дефектний ген. Саме тому родинні шлюби в багатьох країнах заборонені.
Якщо розглядати випадок народження дітей в звичайній родині, то у носіїв такої мутації вони можуть бути:
- 25% ймовірності, що дитина народиться хворою.
- У 50% випадків маля здорове, але є носієм дефектного гена.
- Четверта частина потомства буде абсолютно нормальною.
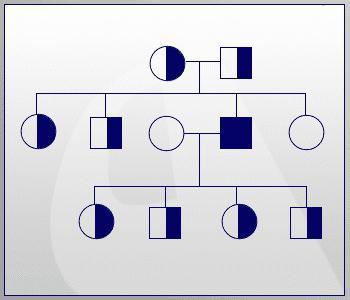
Ця схема не дає повної картини народжуваності хворих дітей. Вона тільки відображає ймовірність, тому у кожної сімейної пари відсоток дефектних генів може бути свій, і передбачити результат, на жаль, неможливо. Зараз існують консультації, в яких фахівці-генетики допомагають парам спрогнозувати народження у них хворої дитини, розповідаючи при цьому, як фенілкетонурія успадковується.
лікування
Як тільки дитині поставлений такий діагноз, заходи необхідно приймати негайно. В першу чергу, потрібно виключити з раціону білкові продукти. Дотримуватися таке суворе обмеження необхідно до 10-12 років, а краще і все життя.
Так як немовлята знаходяться на грудному вигодовуванні і зазвичай нічого, крім материнського молока, ще не вживають, то лікарі рекомендують мамі скоротити його споживання дитиною. Зробити це можливо тільки за однієї умови: давати малюкові зціджене молоко, щоб точно бачити його кількість.
Догодовувати доведеться сумішами, які в своєму складі не мають фенілаланіну. Коли настане час введення прикорму, необхідно вибирати пюре і каші без додавання молока. Можна давати соки, овочеві пюре.
Лікар обов’язково призначає і медикаментозне лікування. Зазвичай це препарати, які містять фосфор, адже цей елемент не дарма вважають «елементом життя і думки», так як він грає важливу роль в роботі нашого мозку. Також призначаються ліки, що містять залізо, кальцій, вони допомагають поліпшити кровообіг і мозкову діяльність.
Лікування не повинно зводитися до повного виключення фенілаланіну, так як в цьому випадку може спостерігатися його недолік, що призводить до занепаду сил, втрати апетиту. Крім того, починається пронос і виникають висипання на шкірі.
Щоб дізнатися, наскільки лікування ефективне, слід періодично здавати аналіз крові і сечі на вміст фенілаланіну.
Захворювання у дітей
Саме в дитячому віці організм розвивається такими темпами, яких не буде в інші періоди життя. Тому в цей час важливо вжити всіх заходів для нормального розвитку нервової системи. Діти, хворі на фенілкетонурію, потребують не тільки в прийомі медичних препаратів і спеціального харчування, а й особливому до себе ставлення.
Перш за все, це постійна увага, щоб від пильного ока матерів не сховалися найменші відхилення в розвитку. Можна застосовувати такі методи лікування:
- лікувальна фізкультура, яка допоможе дитині нормально розвиватися фізично;
- масаж;
- психологічна допомога;
- корекційна педагогіка.

Батьки повинні розуміти, що життя і здоров’я їхньої дитини буде більшою мірою залежати від них самих. Яку обстановку вони зможуть створити навколо хворого малюка, наскільки точно будуть дотримані рекомендації лікарів по харчуванню, чи будуть близькі люди реагувати на відхилення в розумовому і фізичному розвитку – всі ці моменти дуже важливі.
Народна медицина для позбавлення від недуги
Народні рецепти знаходять своє застосування при лікуванні багатьох хвороб. Не виняток і фенілкетонурія. Що це захворювання вимагатиме перегляду всього способу життя – це факт. Дитина повинна рости і мати уявлення про свою недугу.
Батьки зобов’язані в доступній формі пояснити йому, коли він буде здатний усвідомлювати отриману інформацію, наскільки серйозно його положення. Дієту і лікування необхідно дотримуватися протягом усього життя.
Тільки в цьому випадку можна гарантувати повноцінне існування.
Народні лікарі при фенілкетонурії рекомендують вживати більше рослинних білків. У такій їжі фенілаланіну набагато менше, ніж в тваринних продуктах. Чи не забороняється в раціон вводити фрукти і овочі.
У них багато вітамінів і мікроелементів, які в обов’язковому порядку необхідні для нормальної роботи нервової системи.
Тобто народна медицина дотримується думки, що такому хворому бажано дотримуватися вегетаріанську дієту.
Харчування при фенілкетонурії
Фенілаланін міститься практично у всіх продуктах, в яких є білок. Необхідно постаратися виключити їх зі свого раціону, і в першу чергу це стосується молока і м’яса.
Якщо поставлений діагноз «фенілкетонурія», харчування має бути переглянуто в першу чергу. Всі продукти при цьому можна розділити на кілька груп:
- Дозволені до використання завжди: картопля, цукор, чай, рослинні масла.
- Можна вживати в невеликій кількості: рис, мед, масло вершкове, хлібобулочні вироби, овочі і фрукти.
Повністю необхідно виключити зі свого меню: яйця, рибу і м’ясо, молоко, макарони, бобові, горіхи, кукурудзу, молочні продукти, шоколад.

З огляду на той факт, що фенілаланін перетворюється в здоровому організмі в тирозин, то хворі на фенілкетонурію повинні в раціон включати продукти, що містять його в достатній кількості. До такої їжі можна віднести гриби і рослинні складові.
Прогноз на майбутнє при фенілкетонурії
Очевидно, що дане захворювання вимагає негайного вжиття заходів, в іншому випадку життя людини буде короткою.
Захворювання «фенілкетонурія» вимагає уважного ставлення до пацієнта. Якщо дотримуватися суворої дієти і виконувати всі рекомендації лікарів, то дитина зможе нормально рости і розвиватися. Прогноз буде також залежати від того, які захворювання супроводжують генетичному недугу і чи є інші патології.
Поступово, з віком, організм може в якійсь мірі пристосовуватися до підвищеного вмісту фенілаланіну, тому можна іноді допускати послаблення в дієті. Головне, щоб не захопитися цими слабкостями і вчасно зупинитися і перейти на правильне харчування.
https://www.youtube.com/watch?v=PKmqd-OIMjY
Якщо жінка страждає на цю недугу, то їй доведеться ще суворіше ставитися до дотримання всіх рекомендацій, адже вона – майбутня мама. Тільки в цьому випадку вона може народити здорову дитину.
Це особливо актуально, якщо враховувати, що методів профілактики даного захворювання практично не існує.
Фенілкетонурія: симптоми і лікування
Фенілкетонурія (фенилпировиноградная олігофренія, хвороба Фелінга) – це спадкове захворювання, пов’язане з порушенням обміну амінокислоти фенілаланіну. В результаті накопичення токсичних продуктів через неправильне метаболізму розвивається відставання в розумовому і фізичному розвитку.
Причому з’явилися порушення в стані здоров’я незворотні, але своєчасна діагностика захворювання може запобігти всім патологічні зміни. Лікування полягає у виключенні продуктів харчування, що містять фенілаланін. Якщо така елімінаційна дієта застосовується практично з народження, людина виростає здоровим.
Давайте дізнаємося детальніше, що ж це за захворювання, ніж воно проявляється, як діагностується і лікується.
Вперше описана в 1934 р доктором Фелінга, звідки і отримала свою другу назву. Зустрічається з частотою в середньому 1: 10 000, але в різних країнах світу існують коливання від 1 2600 (в Туреччині) до 1: 100 000 (в Фінляндії і Японії), в України від 1: 5 000 до 1:10 000.
причини фенілкетонурії
В основі захворювання лежить генетичний дефект – мутація гена 12-ї хромосоми (98% всіх випадків фенілкетонурії). Це так звана класична фенілкетонурія.
Ген кодує кількість ферменту фенілаланін-4-гідроксилази. Фермент відповідає за перетворення амінокислоти фенілаланіну в організмі людини в тирозин в клітинах печінки.
Фенілаланін – це амінокислота, яка міститься в білкових продуктах (м’ясо, риба, молоко, яйця та інші).
- При мутації гена кількість ферменту знижується, що призводить до накопичення фенілаланіну і продуктів проміжного метаболізму в тканинах організму. Організм намагається позбутися від фенілаланіну і продуктів його розпаду, виводячи їх з сечею.
- Подібні порушення обміну речовин призводять до порушення будови нервових провідників, зниження утворення нейромедіаторів. Все це, поряд з прямим токсичною дією надлишку фенілаланіну, призводить до розвитку розумових порушень, які є основним проявом захворювання.
- Фенілкетонурія успадковується по аутосомно-рецесивним типом, тобто не залежить від статі, і виникає при збігу двох патологічних генів від батька і матері.
Решта 2% випадків фенілкетонурії пов’язані з іншими генетичними дефектами і залежать від концентрації інших ферментів (дігідроптерідінредуктази і ін.).
Вони мають ті ж клінічні прояви, але не піддаються лікуванню дієтою. Такі варіанти відносять до атипового перебігу захворювання. Серед них прийнято виділяти фенілкетонурію II і III.
Генетичний дефект при фенілкетонурії II розташовується в 4-й хромосомі, при III – в 11-й хромосомі.
симптоми
 У дітей з фенілкетонурією світле волосся, бліда шкіра і блакитні очі.
У дітей з фенілкетонурією світле волосся, бліда шкіра і блакитні очі.
Дитина з фенілкетонурією народжується зовні здоровим, тобто нічим не відрізняється від інших дітей. З надходженням їжі в організм починається потрапляння білка, а значить і фенілаланіну. Останній поступово накопичується, і зазвичай до 2 місяців життя з’являються перші симптоми: млявість або неспокій, відсутність інтересу до навколишнього світу, відрижки, зміни м’язового тонусу. Іноді відрижки настільки часті і рясні, що виникає підозра на патологію шлунково-кишкового тракту (пілоростеноз). Дитина через зригування може погано набирати у вазі.
- До 4-6 місяців стає очевидною затримка психічного розвитку. Дитина не стежить за іграшкою, не реагує на звук, не впізнає батьків.
- Чим довше триває надходження фенілаланіну в організм з їжею, тим виражено порушення в психічній і розумової сферах. Розвиток мови різко затримується. Іноді словниковий запас може обмежуватися кількома словами.
- Якщо діагноз не буде виставлений і не буде розпочато лікування, то до 3-4 років розумові порушення досягнуто ступеня ідіотії (найважча ступінь олігофренії).
- Особливістю клінічного перебігу фенілкетонурії є незворотність виникли психічних і інтелектуальних змін. Тобто при пізньому виявленні допомогти таким діткам вже не можна – на все життя вони залишаються розумово відсталими.
Фізичний розвиток також відстає: діти пізніше починають тримати голову, перевертатися, сидіти. Коли такі діти починають ходити, то при цьому вони широко розставляють ніжки, згинаючи їх одночасно в колінних і тазостегнових суглобах.
Хода погойдується, дрібними кроками. У положенні сидячи діти приймають «позу кравця» – згинають і руки, і ноги, підгортаючи останні під себе. Зазвичай обсяг голови менше, ніж в нормі. Може бути виражена мікроцефалія: маленька голова.
З інших неврологічних симптомів можливі порушення м’язового тонусу, судомні напади. Епілептичні напади зазвичай з’являються у віці 1,5 років і призводять до ще більшого прогресування порушень інтелекту.
У частини хворих на фенілкетонурію з’являються мимовільні рухи в кінцівках, тремтіння (гіперкінези). У рухах немає плавності і узгодженості, порушується рівновага.
Крім ряду психічних і інтелектуальних змін, фенілкетонурію характеризують такі симптоми:
- специфічний «мишачий» запах (або запах цвілі) від дитини: цей симптом характерний тільки для фенілкетонурії. Запах з’являється в результаті виділення продуктів метаболізму фенілаланіну (фенилпировиноградной, фенілмолочная, фенилуксусной кислот) через шкіру і з сечею;
- шкірні прояви: дерматити, екзема, просто лущення (виникають з тієї ж причини, що і «мишачий» запах);
- пізнє прорізування зубів: у таких дітей перші зуби можуть з’явитися після 18 місяців, емаль недорозвинена;
- порушення пігментації: у таких дітей зазвичай блакитні очі, дуже світла шкіра і волосся в результаті зниження кількості меланіну (його зміст залежить від метаболізму фенілаланіну). Через це у таких дітей спостерігається підвищена чутливість до сонячного світла;
- вегетативні симптоми: знижений артеріальний тиск, підвищена пітливість, запори, акроціаноз (синюшність кистей і стоп);
- нерідко фенілкетонурія супроводжується вродженими вадами серця.
Атипові випадки фенілкетонурії, пов’язані з порушенням діяльності інших ферментів, які беруть участь у метаболізмі фенілаланіну, крім розумових змін характеризуються розвитком м’язової слабкості у всіх кінцівках з одночасним підвищенням м’язового тонусу, спастичних тетрапарезом. Також при цих формах розвивається слинотеча, напади підвищення температури.
У дорослих людей, які страждають на фенілкетонурію, можлива поява судомних нападів, порушень координації, тремтіння в кінцівках, погіршення пам’яті та уваги, виникнення депресії. Зазвичай подібні симптоми виникають при недотриманні елімінаційної дієти.
діагностика
 Скринінгові тести на вміст фенілаланіну всім новонародженим проводять в пологовому будинку.
Скринінгові тести на вміст фенілаланіну всім новонародженим проводять в пологовому будинку.
У зв’язку з тим, що фенілкетонурія супроводжується розвитком незворотних розумових порушень, у багатьох країнах світу, в тому числі і в України, прийнято використовувати скрініга-методи діагностики. Що це означає? Всім без винятку новонародженим дітям в пологовому будинку проводять експрес-тести на вміст фенілаланіну. Для цього беруть капілярну кров (з п’яти) на 4-5-й день життя дитини (у недоношених на 7-й), наносять на спеціальний паперовий бланк і відправляють в лабораторію, де за певних змін лікар-лаборант робить висновки про зміст фенілаланіну в крові. Негативний тест говорить про відсутність фенілкетонурії.
Якщо тест виявляється позитивним, то тоді проводять додаткові дослідження для визначення вмісту фенілаланіну в крові і сечі (хроматографію, флюоріметре). Концентрацію фенілаланіну в крові і сечі регулярно перевіряють при проведенні лікування, щоб контролювати ефективність дієти і коригувати її при необхідності.
Можливе проведення генетичного дослідження для підтвердження мутації в гені, відповідальному за фенілаланін-4-гідроксилази.
Подібне дослідження можливо в якості пренатальної діагностики, тобто на етапі вагітності (беруть навколоплідні води шляхом пункції).
Це інвазивне дослідження роблять за суворими показаннями (наприклад, наявність хворого на фенілкетонурію дитини в сім’ї). Виявлення генетичного дефекту у плода дозволяє перервати вагітність.
лікування фенілкетонурії
На сьогоднішній день найефективнішим і найпоширенішим способом лікування фенілкетонурії є елімінаційна дієта: дієта з виключенням продуктів, що містять фенілаланін. Якщо її строго дотримуватися в перші роки життя дитини, коли розвиток нервової системи ще триває, то можна виростити здорового і повноцінного людини.
Дуже важливо виняток фенілаланіну саме в перший рік життя, коли найбільш активно розвивається нервова система. Якщо елімінаційна дієта призначається після року, розумові порушення не виліковуються. Щомісяця першого року життя без застосування дієти обходиться дитині безповоротною втратою близько 4 балів IQ.
- Зазвичай достатньо дотримуватися дієти до 16-18 років, після цього віку організм стає менш чутливим до токсичної дії фенілаланіну, і можливе розширення раціону харчування. Включення нових продуктів необхідно проводити під контролем вмісту фенілаланіну в крові. Іноді потрібно довічне суворе дотримання дієти.
- Вагітним жінкам і жінкам, які планують вагітність, і при цьому хворим на фенілкетонурію, для народження здорової дитини обов’язково суворе дотримання дієти.
- Ступінь суворості дієти залежить від концентрації фенілаланіну в крові у дитини. При його рівні до 2-6 мг% (120-360 мкмоль / л) дієта не призначається, вище цього показника – обов’язкова.
- Суть дієти полягає у виключенні білкових продуктів.
Відмова від грудного вигодовування не обов’язковий, але в цьому випадку мати грудної дитини повинна строго дотримуватися елімінаційної дієти, тому що грудне молоко містить білок (відповідно і фенілаланін). Питання про можливість грудного вигодовування вирішується індивідуально !!!

У України забезпечення лікувальним харчуванням дітей, хворих на фенілкетонурію, згідно із законом безкоштовне.
Хворим на фенілкетонурію протипоказані такі продукти: м’ясо, риба (і морепродукти), горіхи, сир, твердий сир, бобові, яйця, вироби з пшеничного борошна, гречана і манна крупа, вівсяні пластівці.
Під час призначення елімінаційної дієти необхідний суворий контроль вмісту фенілаланіну в крові: перші 3 місяці життя – щотижня, від 3-х місяців до року – мінімум раз на місяць, від року до 3-х років – 1 раз в 2 місяці. Прагнуть до змісту фенілаланіну 2-6 мг% у молодших дітей, після 10 років – до 10 мг%. Обов’язково спостереження у дитячого психоневролога.

Атипові форми фенілкетонурії не піддаються лікуванню елімінаційної дієтою. У цьому випадку показано застосування гепатопротекторів, антиконвульсантів, препаратів з леводопою (для корекції гіперкінезів), 5-оксітріптофана, тетрагідробіоптеріна (ВН 4). Ці форми фенілкетонурії мають гірший прогноз для життя і тим більше інтелектуального розвитку.
На сьогоднішній день розробляються нові напрямки в лікуванні фенілкетонурії. Серед них варто відзначити наступні:
- використання замісної терапії фенілаланінліазой (PAL) – рослинним ферментом, що розщеплює фенілаланін до нетоксичних сполук;
- генна інженерія (введення штучно створеного нормального гена, відповідального за фенілаланін-4-гідроксилази);
- метод «великих нейтральних амінокислот» – зменшення всмоктування фенілаланіну з їжі і надходження в головний мозок за допомогою спеціальних препаратів.
Поки ці сучасні розробки не мають широкого застосування, але деякі дослідження, що підтверджують їх ефективність, вже проводяться.
материнська фенілкетонурія
Якщо жінка, яка страждає на фенілкетонурію, при плануванні вагітності і при її настання не дотримується дотримання дієти, то це відбивається на розвитку її дитини. Потомство таких жінок має затримку внутрішньоутробного розвитку та вроджені вади розвитку: пороки серця, аномалії розвитку головного мозку, сечового міхура, мікроцефалія, аномалії лицьового скелета (ущелини).
Щоб запобігти патологічні зміни у дитини, таким жінкам необхідно дотримуватися елімінаційної дієти до зачаття і всю вагітність. Дефіцит білка заповнюється за рахунок спеціальних білкових сумішей без фенілаланіну.
Таким чином, фенілкетонурія – це генетичне порушення амінокислотного обміну, яке при пізній діагностиці призводить до розвитку виражених інтелектуальних порушень у дитини.
Скринінг цього захворювання в пологових будинках дозволяє діагностувати його в перші тижні життя і вчасно призначити лікування.
Основним методом в даний час є призначення елімінаційної дієти, яка дозволяє зберегти інтелект маленькій людині, а значить, зберегти здоров’я, що забезпечить повноцінне існування протягом усього життя.
Фенілкетонурія – що це за захворювання, чому воно виникає, і як лікувати дитину?

Дізнавшись, що це за захворювання – фенілкетонурія, діагностування якого проводиться ще в період новонародженості, при його виявленні потрібно негайно приступити до початку лікування. Раннє виявлення і терапія дозволяють досягти сприятливих результатів.
Фенілкетонурія – що це за хвороба?
Фенілкетонурія, або хвороба Фелінга, є важкою патологією, вперше описаної в 1934 році норвезьким вченим Фелінга.
Тоді Феллінг провів обстеження декількох дітей з розумовою відсталістю і виявив у них присутність в сечі фенілпірувата – продукту розпаду надходить з їжею амінокислоти феніланіну, яка в організмі хворих не розщеплюється.
Фенілкетонурія – захворювання, пов’язане з порушенням обміну речовин вродженого характеру, відкрите одним з перших.
Фенілкетонурія – тип спадкування
Хвороба Фелінга є хромосомно-генетичної, спадкової, що передається дітям від батьків. Винуватцем розвитку патології виступає ген, що знаходиться на 12 хромосомі. Він відповідальний за виробництво печінкового ферменту фенілаланін-4-гідроксилази, за рахунок якого відбувається перетворення фенілаланіну в іншу речовину – тирозин (воно потрібно для нормальної роботи організму).
Встановлено, що фенілкетонурія успадковується як рецесивна ознака. Приблизно 2% людей є носіями дефектного гена, але при цьому не страждають на фенілкетонурію.
Патологія розвиває тільки тоді, коли і мати, і батько передають ген дитині, а статися це може з імовірністю 25%.
Якщо фенілкетонурія успадковується як рецесивна ознака, дружина гетерозиготна, а чоловік гомозиготен по нормальному аллелю гена, то ймовірність того, що діти будуть здоровими носіями гена фенілкетонурії, дорівнює 50%.
форми фенілкетонурії
Розглядаючи, у кого може розвинутися фенілкетонурія, що це за захворювання, найчастіше мова ведеться про класичній формі патології, яка зустрічається приблизно в 98% випадків.
Решта випадків – кофакторная фенілкетонурія, обумовлена дефектом тетрагідробіоптеріна внаслідок порушення його синтезу або відновлення активної форми.
Дана речовина служить кофактором ряду ферментів, і без нього неможливо прояв їх активності.

Фенілкетонурія – причини
Хвороба Фелінга є патологією, при якій через мутацій в гені, що викликають дефіцит або відсутність фенілаланін-4-гідроксилази, відбувається накопичення в тканинах і фізіологічних рідинах фенілаланіну, а також продуктів його неповного розщеплення. Частина надлишкового фенілаланіну перетворюється в фенілкетони, що виводяться із сечею, що і зумовило назву хвороби.
Порушення обмінних процесів позначається більшою мірою на головному мозку. На його тканини проводиться отруйну дію, порушуються процеси жирового обміну, відбувається збій мієлінізації нервових волокон, зниження утворення нейромедіаторів. Так починається запуск патогенетичних механізмів затримки розумового розвитку у дитини.
Фенілкетонурія – симптоми
При народженні дитина з цим діагнозом виглядає здоровим, і тільки через 2-6 місяців виявляються перші симптоми. Фенілкетонурія ознаки починає проявляти тоді, коли в дитячому організмі накопичується фенілаланін, що надходить разом з грудним молоком або сумішами для штучного вигодовування. Можуть відзначатися такі, поки ще не специфічні симптоми:
Крім цього, хворі малюки мають світліший шкірний покрив, волосся і очі, ніж у здорових членів сім’ї, що пов’язано з порушенням вироблення в організмі пігменту меланіну. Ще один діагностична ознака, який можуть помітити медики або уважні батьки, – своєрідний «мишачий» запах, викликаний виділенням феніланіну з сечею і потом.
Більш явними клінічні прояви стають приблизно в піврічному віці, після введення першого прикорму:
- нездатність фокусування погляду на окремих предметах;
- байдужість щодо всього, що відбувається;
- відсутність міміки, посмішки;
- тремор рук і так далі.
Впадають в око й фізичні відхилення: маленький розмір голови, видатна вперед верхня щелепа, відставання в рості. Хворі дітки пізно починають тримати голову, повзати, сідати, вставати. Характерна особлива поза в положенні сидячи – поза «кравця», при якій ручки постійно зігнуті в ліктях, а ніжки – в колінах. У віці трьох років, якщо лікування так і не розпочато, симптоматика наростає.
Фенілкетонурія – діагностика
Фенілкетонурія у дітей виявляється найчастіше ще в пологовому будинку, що дозволяє вчасно почати лікування і не допустити розвитку ряду незворотних наслідків.
На 4-5 добу після народження у малюків беруть капілярну кров натщесерце для визначення деяких важких генетичних захворювань, серед яких – фенілкетонурія.
Якщо виписка з пологового будинку сталася раніше, аналіз роблять в поліклініці за місцем проживання протягом перші 10 діб життя.
З урахуванням того, що в окремих випадках бувають помилкові результати, діагноз ніколи не встановлюється за підсумками першого аналізу. Для підтвердження наявної патології призначається ряд інших досліджень, серед яких:
- аналіз сечі на виявлення фенілпірувата;
- кількісне визначення природних амінокислот в плазмі;
- визначення активності печінкових ферментів;
- електроенцефалографія і магнітно-резонансна томографія головного мозку.
Генетичний дефект, який призводить до розвитку патології, може бути виявлений у плода при інвазивної пренатальної діагностики. Для цього відбираються проби клітин з ворсинок хоріона або амніотичної рідини, а потім проводиться ДНК-аналіз. Рекомендується таке дослідження в сім’ях з високим ризиком захворюваності, в тому числі, якщо вже є дитина з фенілкетонурією.
Фенілкетонурія – лікування
Коли виявляється фенілкетонурія у новонароджених, обов’язково хворих повинні спостерігати лікарі таких спеціальностей, як генетик, педіатр, невропатолог, дієтолог.
Тим, кому відомо, фенілкетонурія – що це за захворювання, буде зрозуміло, чому основою його лікування є дотримання дієти з обмеженням фенілаланіну.
Крім того, призначається медикаментозне лікування, масаж, лікувальна фізкультура, психолого-педагогічні методики для соціалізації дитини, підготовки його до навчання.

Фенілкетонурія – дієта
При постановці діагнозу «фенілкетонурія», дієта для дитини призначається негайно. З раціону виключаються продукти, багаті на білок (м’ясо, риба, молочні продукти, бобові, горіхи та інші).
Потреба в білках відшкодовується за рахунок спеціальних дієтичних сумішей та інших продуктів з берлофеном – полусинтетическим гидролизатом білка, повністю позбавленим фенілаланіну (Тетрафен, Лофеналак, Нофелан).
Хворі вживають безбілковий хліб, макаронні вироби, крупи, муси і так далі. Грудне годування проводять в обмежених дозах.
Найсуворіше дотримання дієти з регулярним контролем вмісту фенілаланіну в крові протягом перших 14-15 років життя запобігає розвитку розумових відхилень.
Далі раціон кілька розширюється, але багато фахівців рекомендують довічне дотримання спеціального раціону.
Кофакторная форма фенілкетонурії за допомогою дієти не лікується, а коригується тільки введенням препаратів тетрагідробіоптеріна.
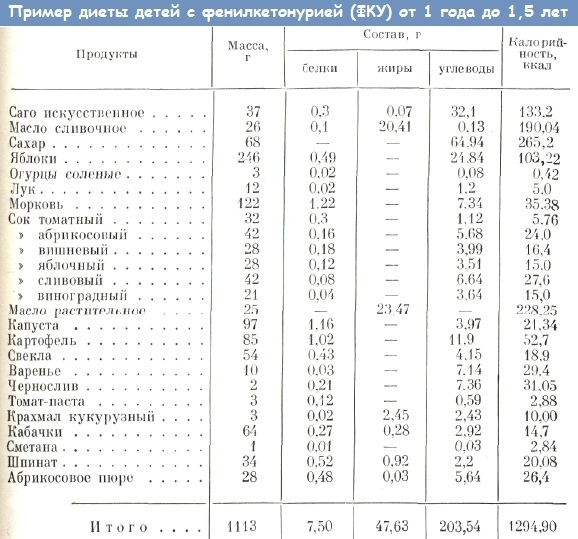
Фенілкетонурія – препарати для лікування
Фенілкетонурія лікування у дітей передбачає і прийом деяких препаратів, серед яких:
- ноотропні засоби (пірацетам, Церебролізин);
- вітаміни групи В;
- мінеральні комплекси;
- препарати для поліпшення тканинного обміну (АТФ, Рибоксин);
- засоби, що поліпшують мікроциркуляцію (трентал, Пентоксифілін).
Фенілкетонурія – прогноз щодо життя і хвороби
Батькам, які не з чуток знають, що це за генетичне захворювання – фенілкетонурія, в сучасних умовах надається можливість виростити здорову дитину, якщо слідувати всім лікарським приписи. Коли належного лікування немає, фенілкетонурія прогноз має невтішний: хворі живуть близько 30 років з важкою розумовою недостатністю і множинними функціональними порушеннями.
Як успадковується фенілкетонурія?
Фенілкетонурія – це важке спадкове захворювання, воно з’являється у дітей, чиї батьки є носіями пошкодженого гена.
В організмі людини, хворого фенікетонуріей через порушення роботи ферментів накопичується фенілаланін, його передозування викликає важкі незворотні пошкодження нервової системи і всього організму.

Головне, що потрібно знати – фенілкетонурія успадковується по аутосомно-рецесивним ознакою . Тепер розберемося, що це таке.
Всі гени діляться на два види – домінантні і рецесивні . Домінантні – ті, які проявляються, рецесивні – ті, які є, але не виявляються.
Так ось, ймовірність народження дитини з фенілкетонурією є і чоловіки і жінки, які обидва є власниками рецесивного, тобто, «сплячого» гена.
Треба сказати, що такий тип спадкування означає, що захворювання може проявитися як у дівчаток, так і у хлопчиків – ймовірність захворювання на фенілкетонурію однакова. Захворювання проявляється не в кожному поколінні.
ДНК кожної людини містить пару генів, від яких залежить правильна переробка ферменту фенілаланін в тирозин і фенілкетон. Можливі наступні їх варіації:
- Обидва гена домінантні . Людина не хворий і не носій.
- Обидва гена рецесивні . Людина хвора.
- Один ген домінантний і один рецесивний . Людина не хвора, але носій.
Отже, два носія випадкової комбінації таких генів, чоловік і дружина, народжують дитину. Які варіанти успадкування генів можливі тут?
- 50% (25% + 25%) – від одного з батьків домінантний і від одного рецесивний ген, дитина – носій мутованого гена, але не хворий сам.
- 25% – від обох батьків домінантний ген, дитина здорова і не є носієм гена (два домінантних гена говорять про те, що людина не передає у спадок аутосомно-рецесивне захворювання).
- 25% – від обох батьків рецесивний ген, дитина хвора на фенілкетонурію.
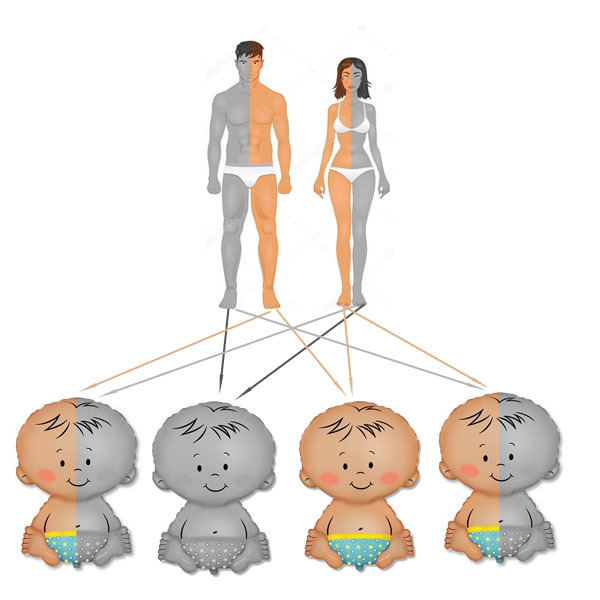
Природно, велика ймовірність того, що обидва батьки матимуть рецесивний ген, виникає в близькоспоріднених відносинах, всередині одного замкнутого поселення, племені і т.д.
- У сучасному світі кожна людина може вибрати собі партнера не тільки зі своїх «сусідів», але скільки часу в історії людства шлюби укладалися саме так – всередині однієї династії, всередині одного села! Ось чому в близькоспоріднених шлюбах народжуються розумово-відсталі діти.
- Так що, кожен з нас може і не хворіти цим захворюванням, але бути потенційним його розповсюджувачем. Якщо у вас є один домінантний і один рецесивний ген, ви носій.
- Якщо у вашого партнера теж один домінантний і один рецесивний ген, то він теж носій.
І якщо вашій дитині від вас передасться рецесивний ген і точно такий же від партнера – дитина народиться з спадковим захворюванням фенілкетонурія.
Так що, хвора дитина може народитися і у здорових батьків .
Чи можна здати аналіз на фенілкетонурію? Як дізнатися чи є людина носієм гена фенілкетонурії ? Де роблять аналізи на фенілкетонурію?
Цілком розумно, що кожна пара, плануючи завести дитину і знаючи про такий небезпечному і складному захворюванні, задається питанням – наскільки ймовірним є народження дитини з фенілкетонурією?
Дізнатися це можна, здавши тест. Зараз в країні дуже багато приватних лабораторій та клінік, потрібно лише знайти серед них ті, де роблять молекулярно-генетичну діагностику. Серед кількох типів аналізу ДНК присутній і діагностика фенілаланінгідроксилази (ФАГ) – того самого гена, пошкодження якого викликає порушення в перетворенні фенілаланіну в тирозин.
ФАГ розташовується в 12 хромосомі і при нормальній роботі виробляє фенілаланін-гідроксилази, яка в свою чергу і допомагає організму перетворити фенілаланін в тирозин. При порушенні роботи ферменту феніаланін не переробляється і накопичується в організмі.
Він надає токсикологічне вплив на організм, це свого роду отрута – він вражає весь організм людини, в першу чергу і особливо сильно – нервову систему.
Накопичуючись в крові, феніаланін практично не виходить з організму, лише невелика його кількість може бути виведено разом з сечею, яка, до речі, набуває характерний запах, але цього недостатньо.
- Втім, все про фенілкетонурії ви можете дізнатися з нашої статті.
- Додамо, що в даний час аналіз на фенілкетонурію є обов’язковим і його роблять абсолютно всім немовлятам в пологовому будинку під час скринінгу – забору крові з п’ятки і аналізу її на майже півсотні різних захворювань, як спадкових, так і немає.
фенілкетонурія
Фенілкетонурія – спадкове порушення амінокислотного обміну, обумовлене недостатністю печінкових ферментів, що беруть участь у метаболізмі фенілаланіну до тирозину. Ранніми ознаками фенілкетонурії служать блювота, млявість або гіперактивність, запах цвілі від сечі і шкіри, затримка психомоторного розвитку; типові пізні ознаки включають олігофренію, відставання у фізичному розвитку, судоми, екзематозні зміни шкіри та ін. Скринінг новонароджених на фенілкетонурію проводиться ще в пологовому будинку; подальша діагностика включає молекулярно-генетичне тестування, визначення концентрації фенілаланіну в крові, біохімічний аналіз сечі, ЕЕГ, МРТ головного мозку. Лікування фенілкетонурії полягає в дотриманні спеціальної дієти.
Фенілкетонурія (хвороба Фелінга, фенилпировиноградная олігофренія) – вроджена, генетично обумовлена патологія, що характеризується порушенням гидроксилирования фенілаланіну, накопиченням амінокислоти і її метаболітів в фізіологічних рідинах і тканинах з подальшим тяжким ураженням ЦНС. Фенілкетонурія вперше описана А. Фелінга в 1934 р
; зустрічається з частотою 1 випадок на 10 000 новонароджених. У неонатальному періоді фенілкетонурія не має клінічних проявів, однак надходження фенілаланіну з їжею викликає маніфестацію захворювання вже в першому півріччі життя, а в подальшому призводить до важких порушень розвитку дитини.
Саме тому Пресимптоматичне виявлення фенілкетонурії у новонароджених є найважливішим завданням неонатології, педіатрії і генетики.
Фенілкетонурія є захворюванням з аутосомно-рецесивним характером успадкування. Це означає, що для розвитку клінічних ознак фенілкетонурії дитина повинна успадкувати по одній дефектної копії гена від обох батьків, які є гетерозиготними носіями мутантного гена.
Найчастіше до розвитку фенілкетонурії призводить мутація гена, що кодує фермент фенілаланін-4-гідроксилази і розташованого на довгому плечі 12 хромосоми (локус12q22-q24.1).
Це, так звана, класична фенілкетонурія I типу, складова 98% всіх випадків захворювання. Гіперфенілаланінемія може досягати 30 мг% і вище.
При відсутності лікування даний варіант фенілкетонурії супроводжується глибокою розумовою відсталістю.
Крім класичної форми, розрізняють атипові варіанти фенілкетонурії, що протікають з тієї ж клінічною симптоматикою, але не піддаються корекції дієтотерапією. До них відносяться фенілкетонурія II типу (недостатність дегідроптерінредуктази), фенілкетонурія III типу (дефіцит тетрагідробіоптеріна) та інші, більш рідкісні варіанти.
Імовірність народження дитини, хворої на фенілкетонурію, підвищується при укладанні близькоспоріднених шлюбів.
В основі класичної форми фенілкетонурії лежить недостатність ферменту фенілаланін-4-гідроксилази, який бере участь в конверсії фенілаланіну в тирозин в мітохондріях гепатоцитів.
У свою чергу, похідний тирозину – тирамін є вихідним продуктом для синтезу катехоламінів (адреналіну і норадреналіну), а дийодтирозин – для освіти тироксину.
Крім цього, результатом метаболізму фенілаланіну служить утворення пігменту меланіну.
Спадкова недостатність ферменту фенілалаіін-4-гідроксилази при фенілкетонурії призводить до порушення окислення фенілаланіну, що надходить з їжею, в результаті чого його концентрація в крові (фенілаланінемія) і спинномозкової рідини значно зростає, а рівень тирозину відповідно падає. Надлишковий вміст фенілаланіну усувається шляхом підвищеної екскреції з сечею його метаболітів – фенилпировиноградной, фенілмолочная і фенилуксусной кислот.
- Порушення обміну амінокислот супроводжується порушенням мієлінізації нервових волокон, зниженням утворення нейромедіаторів (дофаміну, серотоніну та ін.), Що запускають патогенетичні механізми затримки розумового розвитку і прогредиентное недоумство.
- Новонароджені з фенілкетонурією не мають клінічних ознак захворювання. Зазвичай маніфестація фенілкетонурії у дітей відбувається у віці 2-6 місяців.
- З початком годування в організм дитини починає надходити білок грудного молока або його замінників, що призводить до розвитку перших, неспецифічних симптомів – млявості, іноді – занепокоєння і гіперзбудливості, відрижки, м’язової дистонії, судомного синдрому.
Одним з ранніх патогномонічних ознак фенілкетонурії служить сильна блювота, яка нерідко помилково розцінюється як прояв пілоростеноза.
До другого півріччя стає помітним відставання дитини в психомоторному розвитку. Дитина стає менш активним, байдужим, перестає впізнавати близьких, не намагається сідати і вставати на ніжки. Аномальний склад сечі і поту обумовлюють характерний «мишачий» запах (запах цвілі), що виходить від тіла. Часто спостерігається лущення шкіри, дерматити, екзема, склеродермія.
У дітей з фенілкетонурією, які не отримують лікування, виявляється мікроцефалія, прогнатия, пізніше (після 1,5 років) прорізування зубів, гіпоплазія емалі. Відзначається затримка мовного розвитку, а до 3-4 років виявляється глибока олігофренія (ідіотія) і практично повна відсутність мовлення.
Діти з фенілкетонурією мають диспластичне статура, нерідко – вроджені вади серця, вегетативні дисфункції (пітливість, акроціаноз, артеріальну гіпотонію), страждають запорами.
До фенотипическим особливостям дітей, які страждають на фенілкетонурію, слід віднести світлу шкіру, очі і волосся.
Для дитини з фенілкетонурією характерні специфічна поза «кравця» (зігнуті в суглобах верхні і нижні кінцівки), тремор рук, хитка, семенящая хода, гіперкінези.
Клінічні прояви фенілкетонурії II типу характеризуються важким ступенем розумової відсталості, підвищеною збудливістю, судомами, спастичних тетрапарезом, сухожильной гиперрефлексией. Прогресування захворювання може призводити до загибелі дитини у віці 2-З років.
При фенілкетонурії III типу розвивається тріада ознак: мікроцефалія, олігофренія, спастичний тетрапарез.
В даний час діагностика фенілкетонурії (а також галактоземии, вродженого гіпотиреозу, адрено-генітального синдрому і муковісцидозу) входить в програму неонатального скринінгу, здійснюваного всім новонародженим.
Скринінг-тест проводиться на 3-5 день життя доношеної і 7 день життя недоношеної дитини шляхом забору зразка капілярної крові на спеціальний паперовий бланк. При виявленні гіперфенілаланеміі більше 2,2 мг% дитину направляють до дитячого генетику для повторного обстеження.
Для підтвердження діагнозу фенілкетонурії перевіряється концентрація природних амінокислот в крові, визначають активність печінкових ферментів (фенілаланінгідроксилази), виконується біохімічне дослідження сечі (визначення кетонових кислот), метаболітів катехоламінів в сечі і ін. Додатково проводиться ЕЕГ і МРТ головного мозку, огляд дитини дитячим неврологом.
Генетичний дефект при фенілкетонурії може бути виявлений ще на етапі вагітності в ході інвазивної пренатальної діагностики плоду (хоріонбіопсіі, амніоцентезу, кордоцентеза).
Диференціальний діагноз фенілкетонурії проводять з внутрішньочерепної родовою травмою новонароджених, внутрішньоутробними інфекціями, іншими порушеннями обміну амінокислот.
Основним фактором в лікуванні фенілкетонурії є дотримання дієти, яка обмежує надходження білка в організм. Лікування рекомендується починати при концентрації фенілаланіну> 6 мг%. Для грудних дітей розроблені спеціальні суміші – Афенілак, Лофенілак; для дітей старше 1 року – Тетрафен, Феніл-фрі; старше 8 років – Максамум-ХР і ін.
Основу дієти складають низькобілковий продукти – фрукти, овочі, соки, білковий гідролізат і амінокислотні суміші. Розширення дієти можливо після 18 років у зв’язку зі зростанням толерантності до фенілаланіну. Відповідно до українського законодавства забезпечення осіб, які страждають на фенілкетонурію, лікувальним харчуванням, повинна здійснюватися безкоштовно.
Хворим призначається прийом мінеральних сполук, вітамінів групи В та ін .; за показаннями – ноотропні засоби, антиконвульсанти. У комплексній терапії фенілкетонурії широко використовується загальний масаж, ЛФК, голкорефлексотерапія.
Діти, які страждають на фенілкетонурію, знаходяться під наглядом дільничного педіатра і психоневролога; нерідко потребують допомоги логопеда і дефектолога. Необхідний ретельний моніторинг нервово-психічного статусу дітей, контроль рівня фенілаланіну в крові і показників електроенцефалограми.
Атипові форми фенілкетонурії, що не піддаються лікуванню дієтою, вимагають призначення гепатопротекторів, протисудомних засобів, замісної терапії леводопою, 5-гідроксітріптофаном.
Проведення масового скринінгу на фенілкетонурію в неонатальному періоді дозволяє організувати ранню дієтотерапію і запобігти тяжким церебральні ушкодження, порушення функції печінки. При ранньому призначенні елімінаційної дієти при класичній фенілкетонурії прогноз розвитку дітей хороший. При пізно почате лікування прогноз щодо розумового розвитку несприятливий.
Профілактика ускладнень фенілкетонурії полягає в проведенні масового скринінгу новонароджених, раннього призначення і тривалого дотримання дієтичного харчування.
З метою оцінки ризику народження дитини з фенілкетонурією попереднє генетичне консультування повинні пройти подружні пари, які вже мають хвору дитину, що складаються в кровноспоріднених шлюби, які мають родичів з даним захворюванням.
Жінки з фенілкетонурією, які планують вагітність, повинні дотримуватися суворої дієти до зачаття і під час вагітності для виключення підвищення рівня фенілаланіну і його метаболітів і порушення розвитку генетично здорового плоду.
Ризик народження дитини з фенілкетонурією у батьків-носіїв дефектного гена, становить 1: 4.